9.1 Pdb output settings - Dealing with limitation and compatibility issues
The pdb format is very complex and so are the problems that occur when trying to build a file that syntactically complies with all further needs, in particular when it comes to larger molecule sizes.
Press 'Manage Restrictions'  inside the
'File' menu or the toolbar to open the following dialog:
inside the
'File' menu or the toolbar to open the following dialog:
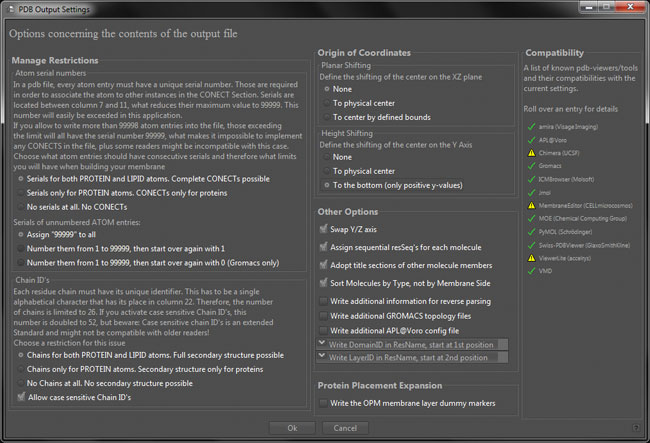
The pdb output settings dialog
This window provides options to manage restrictions, options regarding coordinate transformations and some other settings to be made. The right side displays the compatibility with several known tools. A yellow warning sign indicates that there might be problems with the referring application and the current settings. Roll over it with your mouse in order to get more information.
Manage Restrictions
The pdb-format was originally intended to describe one single molecule per file.
Usually, all of its atoms are serially numbered by integers (consisting of a maximum of five digits), while its chains are identified using single (alphabetical) characters. Due to that, only 99999 different atom serials, plus 26 different chain-id's are possible inside one file.
It was assumed that these capacities would be enough for any molecule. But when we generate a pdb-file that contains an entire membrane, with its vast number of atoms and (eventually) chains, these limits will quickly be exceeded.
By omitting the atom serials, it is actually possible to have more that 99999 atoms in your membrane file. Although it would not be well-formed anymore, most readers will still accept it.
Note that eventual connectivity-annotations will be rejected, since they use the serials to reference specific atoms. Analogically to this, potential chain ids can be also omitted. Without them, the file will even remain well-formed, but the according primary-, and therefore secondary structures cannot be maintained.
However, this issue requires you to choose what contents the membrane file should include and to set the appropriate limits. You can do so any time, regardless of your current project.

In the upper part you can choose what kind of atoms should be taken into account for consecutive numbering: All atoms, atoms of proteins only or none at all. The last choice enables you to have an unlimited number of atoms inside your membrane.
In the lower part you have the same options concerning the different chain-id's: To either use the chain ids of all molecules (although it is very unlikely that lipids contain any chains at all), to regard only the chains in proteins or not to adopt any chain ids at all.
Another option is to allow case-sensitive chain id's. Most readers will accept them, since they are common in the modern pdb-format. Enabling them will double their maximum count to 52.
While you build your membrane, you can always check your total atom and chain counts which are displayed below.

The bars denounce how close you are to both limits. If one limit is exceeded, you will not be able to generate the membrane file, unless you remove enough objects or increase your limitation settings.
The last option here is concerning the serials of unnumbered ATOM entries. This is only relevant if there are any unnumbered ATOM entries found in the PDB file. You can choose whether they should all get the number 99999 or be serially numbered from 0 while starting all over again when 99999 is reached.
The iterative numbering of atom entries restarts usually with 1, because the numbering in a regular PDB file also starts with 1. But there is now a special option which mimics the numbering behavior of GROMACS which restarts the numbering with 0 (instead of 1). However, the first numbering in a GROMACS-based PDB file also runs from 1 to 99999.
Origin of Coordinates
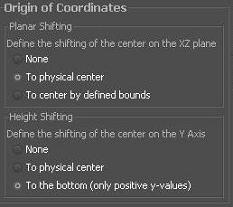
In this section you are able to set up the transformation of all atom points in the file. No translation will leave the zero-point in the upper right hand corner and all extracellular lipids will be in positive while all extracellular lipids will be in negative y-range.
Setting the XZ or Y center to physical will normalize the center point to the "real" middle of all coordinates.
The XZ center can furthermore be set to the center by the membrane bounds defined in this application.
Setting the Y center to the bottom will result in only positive y-values
Other Options

Swap Y/Z Axis:
Swaps the Y and the Z axis. Some coordinate systems in other applications require this
Assign sequential resSeq's for each molecule:
If selected, all resSeq's of one molecule will get the same number, incrementing by 1 at the next molecule. Disabling this option will leave the resSeq's at their original values
Adopt title sections of other molecule members:
Use this feature in order to insert a copy of the title section of each type of molecule that is a member. The title sections will be placed inside the membrane's REMARK field, and not be altered in any way.
Doing this will raise the required disc space. But in some cases it might be necessary to attach information about the included molecules simply to avoid copyright violations.
Sort Molecules by Type,not by Membrane Side:
If this option is selected, the lipids are not sorted by side during exporting. Usually, first all lipids of the extracellular layer, and then the ones of the intracellular level are written to the PDB file(if this option is NOT selected). But if this option is selected, all lipids are sorted by type: first all lipids of type A are written, then all lipids of type B, etc.
Write additional information for reverse parsing:
Activate this option to be able to open the PDB file with the MembraneEditor again. For this purpose, the complete XML file is saved in the REMARK section of the PDB file. Usually, this section is ignored by external programs. But this option is needed to open a PDB file.
 But there is
another alternative. If a PDB file was exported, it is possible to
parse this file again into the MembraneEditor by just indicating the
original PDB file during the loading process.
But there is
another alternative. If a PDB file was exported, it is possible to
parse this file again into the MembraneEditor by just indicating the
original PDB file during the loading process.
Write DomainID and/or LayerID in ResName:
The following line shows a PDB entry of a single C atom in a membrane PDB file:
ATOM 18205 C1 DPP 247 14.438 10.443 64.480 1.00 0.00
The ResName DPP is used here to identify a specific lipid type, DPPC. (The PDB format provides only three digits, therefore it is not possible to use longer names.)
So imagine now the following case: you have a membrane with a microdomain in the center. The default area surrounding the microdomain contains DPPC and also the microdomain contains DPPC. If you export now this membrane to PDB format and you start a simulation with this membrane, it will not be an easy task to judge which lipid was inside the domain and which one outside the domain. For this purpose, this new option was added.
The original PDB file of the lipid does not have to be changed. Just by activate one of the following options the ResName will be combined with the DomainID:
- Write DomainID in ResName, start at 1st position
- Write DomainID in ResName, start at 2nd position
- Write DomainID in ResName, start at 3rd position
For example, here, the option "Write DomainID in ResName, start at 2nd position" was chosen:
ATOM 18205 C1 D0P 247 14.438 10.443 64.480 1.00 0.00
This atom is located in the default area. But this can also be combined with the LayerID by choosing e.g. "Write LayerID in ResName, start at 3rd position":
ATOM 18205 C1 D02 247 14.438 10.443 64.480 1.00 0.00
This atom is located in the default area and in layer 2. If tools like APL@Voro are used now, it will be an easy task to differentiate lipids associated to different membrane areas.
Note: If a membrane contains more microdomains than 9 (plus one default area, number 0), two-digits will be required. In this case, choose the 2nd or 3rd position to insert the DomainID.
Write additional GROMACS topology file:
This option is interesting for those of you who want to simulate the exported PDB file by using external tools like GROMACS. A file topol.top is created which lists all lipid types with the exported molecule numbers depending on the option Sort Molecules by Type,not by Membrane Side. The file will be saved into a folder with the name of the PDB file.
Write additional APL@Voro config file:
If this option is activated, an additonal APL@Voro config file will be generated. This tool is described in section 9.6 Analysis of PDB Membrane Files with APL@Voro. The file will be saved into a folder with the name of the PDB file.
Write the OPM membrane layer dummy markers:
Enable this to write the dummy atoms of potentially included OPM-files into the final membrane file.