7.1. About
This chapter introduces you to the usage of so called 'Molecule Profiles'. Such profiles describe the conformation and even the granularity of a specific molecule, which you can define by yourself. In this way, you are able to apply an alternative conformation and/or granularity to a molecule and so to change the defaults given by the molecule's pdb-file.
Molecule Profiles can be generated, edited and finally applied by the Molecule Editor, which can be accessed by pressing 'Select Profile' in an entry inside the Component View or by pressing 'Manage Molecule Profiles' in the Local Database View:
The Molecule Editor looks the this:
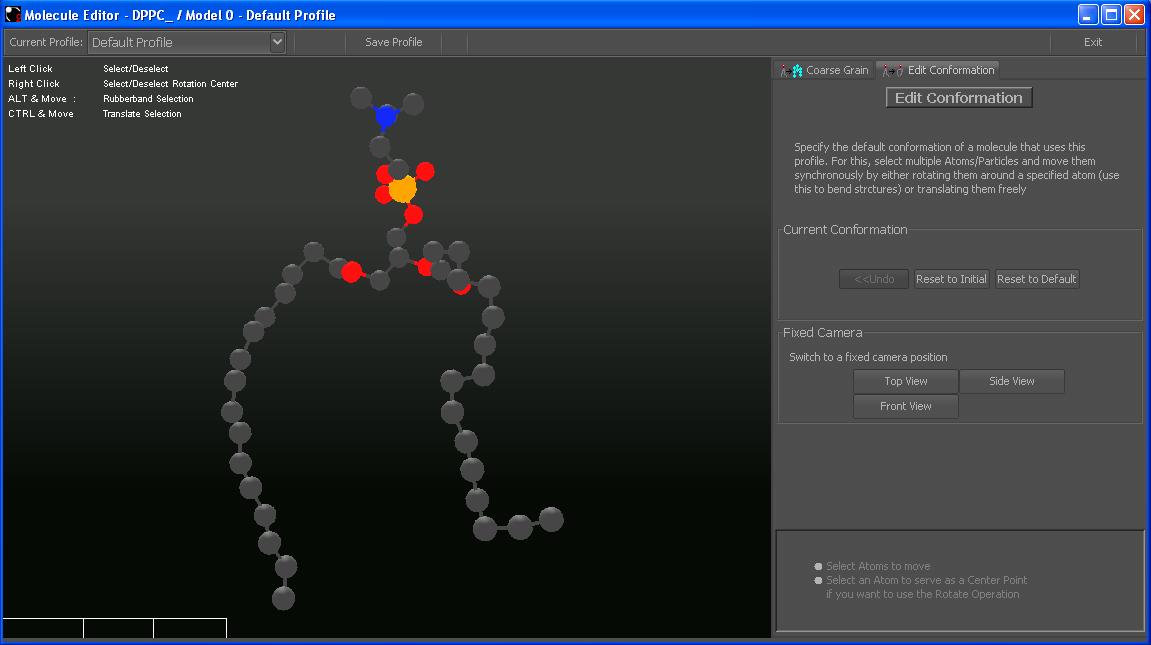
Why edit conformations?
The default conformation, especially of lipids may be quite unpleasant (shown in the picture above). You can either look for a pdb file with a more suitable conformation, or you can edit the conformation here by youself.The advantage is that you can define different conformations in different profiles without needing multiple pdb files.
Why edit the granularity?
This has three reasons:(1) You want to create a membrane that serves as an initial state in further molecular simulation. It could make sense to use this option as a coarse-grain approach, assuming that the simulation is aware of the particles that have been made.
(2) You want to create more simple membranes for simulation and visualization tools, such as the virtual cell, where a reduced number of atoms widely effects the performance (note that fewer atoms, respectively with longer distances between them will still be regarded as bonded if they have been bonded before. Due to the CONECT's written into the final pdb file those bonds remain and should be properly displayed by most pdb viewers).
(3) You can also benefit from simplified molecules if you are using the Atom Level Minimizer algorithm, where the speed of the calculation also depends significantly on the total number of involved atoms.
Managing Profiles
A profile for the viewed molecule can be chosen in the upper toolbar of the Molecule Editor. If any changes have been made, you will also be able to save the current profile or to save it as a new one. Simply use the button 'Save Only' for this task. Enter a new name in order to create a new profile or enter the name of any existing profile to overwrite it. The 'Default Profile' cannot be changed. Stored profiles will always be available for the specific type of molecule, independently of the currently opened Membrane Model.Applying Profiles
This option is only available if the Molecule Editor has been opened from inside the Component View, meaning that you refer to a group of molecules that have already been added to the membrane. By applying a profile, all regarded instances inside the membrane will change their conformation and/or granularity according to the selected profile.Click 'Apply Profile' on the left side in the upper toolbar or 'Save and Apply' if any changes have been made.
You will see another dialog where you have to specify the members that you want to apply to:
