
5.3 Aligning Molecules
What does it mean?
Aligning molecules means that you set the rotation and (for proteins) translate the y-coordinate of its center. This enables it to fit properly into your membrane.
The aligning of both, proteins and sample lipids is performed inside a special mode of the Membrane View. You are looking at the molecule from the side, by default along the membrane's z-axis, in the negative direction. You can change your viewpoint to 5 different angles in the lower right corner of the display and you may enter the free-look-mode in the toolbar to look around without rotating the molecule.
Aligning a Protein/Lipid
To align a protein or lipid, select a single one from the Membrane View and press one of the align buttons in the upper toolbar or in the context menu.
The fist align button  will show the atomic structure of the protein to align together with the neighboring lipids. The number of neighbors is internally computed and depends on the distance between the protein and the lipids as well as the number of atoms.
will show the atomic structure of the protein to align together with the neighboring lipids. The number of neighbors is internally computed and depends on the distance between the protein and the lipids as well as the number of atoms.
By pressing the second align button  the user is able to define the size of the radius in which neighboring molecules are shown. It is also possible to show neighboring proteins and lipids. If both options are deactivated or the size of the radius is set to 0, only the protein to align will be visible. The collision detection with the neighboring molecules will work in any case.
the user is able to define the size of the radius in which neighboring molecules are shown. It is also possible to show neighboring proteins and lipids. If both options are deactivated or the size of the radius is set to 0, only the protein to align will be visible. The collision detection with the neighboring molecules will work in any case.

The Membrane View, while aligning a protein
You will now be able to define a rotation and y-translation for the protein by using the mouse. Hold down the left mouse button to rotate and use the right button to change the y-transformation. The current rotation matrix is displayed in the upper left side of the HUD. The y-translation is displayed in angstrom.
The alignment buttons
In the lower toolbar we have four buttons which can be used to affect the current values:
PDB Alignment
Sets the rotation matrix to a unit matrix, resulting in the same rotation as in the molecule's PDB file.
Flip Protein/Lipid
With this option, the rotation of the protein is inverted. This option will be important, if e.g. different membranes in one file have to be modeled, and the topology of the actual PDB file is inverse to the one proposed by the alignment of OPM or PDBTM (see below).
If this option is chosen for a lipid, the lipid will do a flip flop, which means, it will change the membrane side. But please keep in mind, that the lipid, although it changes the membrane layer, will still be internally associated to the original membrane layer (which means to the Sample Lipids of the original membrane layer).
Enter...
Opens a dialog that enables to define the position and rotation of the molecule manually.
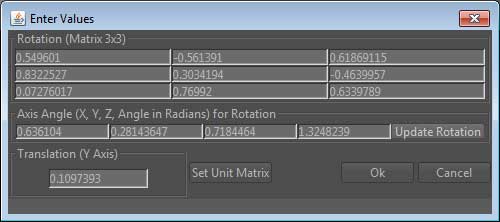
It is possible to define the rotation 3x3 matrix, but keep in mind that its determinant has to be equal to 1. Therefore, it will usually be more appropriate to set the axis angle. The first three values define the X,Y,Z values of the axis, around which the rotation will occur. The fourth value is the angle in Radians. If these value are changed, please do not forget to press the 'Update Rotation' button, which will recompute the new rotation matrix. Finally, it is also possible to change the translation around the Y axis.
Reset
Resets your new alignment to the previous values
Apply to Others
Aligns all other instances of the same type according to your current alignment
Reset Others
Resets the eventually changed alignment of all other instances
OPM Alignment / PDBTM Alignment
Semi-automatic protein alignment
If alignment data of the OPM and/or PDBTM is available for your current protein, you will also find buttons that you can use to apply one of the specific alignments. Because proteins are normally automatically aligned while adding them. This option should only be required if you changed the alignment in the meantime.
 The automatic alignment may be deactivated in
The automatic alignment may be deactivated in
3D Settings -> Controls -> PPE Automatic Protein Placement

An auto-aligned protein. You can see the so called "Dummy Layers" of the OPM file.
When finished, press  to accept your changes, or press
to accept your changes, or press  to discard them.
to discard them.
Topology
The topology for the membrane proteins is as follows, where IN is oriented to the bottom and OUT is oriented to the top in the Alignment Mode:
Plasma membrane:
IN - cytoplasmic,
OUT - extracellular;
Endoplasmic reticulum, Golgi, peroxisome, endosome, vacuole, and vesicle membranes:
IN - cytoplasmic,
OUT - luminal;
Inner bacterial membrane:
IN - cytoplasmic,
OUT - periplasmic or extracellular space;
Outer bacterial membrane:
IN - periplasmic space,
OUT - extracellular;
Inner mitochondrial or chloroplast membrane:
IN - matrix/stroma,
OUT - intermembrane space;
Inner nuclear membrane:
IN - lumen,
OUT - perinuclear space;
Outer mitochondrial, chloroplast or nuclear membrane:
IN - cytoplasmic,
OUT - intermembrane space;
Chloroplast thylakoid membrane:
IN - stromal,
OUT - thylakoid space.
Update and Download
To update the list containing all PDB files supported by PDB_TM or to download OPM files, please refer to 4.1 Search & Download
Aligning custom PDB files
The OPM database provides a relatively new feature: The PPM server.
website: http://opm.phar.umich.edu/server.phpIf you created a protein using prediction tools or if you downloaded a predicted structure which is not found in the PDB database, you can use this server. It will try to create the dummy layers known from the orginal OPM files. This files can be imported into CmME. It will automatically recognice the layers and you can use its semi-automatic alignment feature discussed in this section.
If you want the Local Database to recognize the OPM file and to mark it with the OPM logo, just add the following suffix to your PDB file before you import it to CmME: _OPM (e.g. TestOPMFile1_OPM.pdb). The semi-automatic alignment is not affected by this setting!
Differences between OPM and PDBTM
The topology stated above is also located at the OPM website. It is the newest topology published from both databases. Using OPM files, CmME adjusts the rotation as well as the vertical placement of the protein. Using the PDBTM matrices, only the rotation is adjusted. In many cases this is sufficiant for transmembrane proteins. But sometimes it will be needed to change the vertical translation manually using the methods introduced above.
References
If you use the semi-automatic protein placement feature of CmME, please cite the according publications:
For PDB:
Berman H, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov I, Bourne P (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235-242.
website: http://www.pdb.org
For PDB_TM:
Tusnády GE, Dosztányi Z, Simon I. (2005) PDB_TM: selection and membrane localization of transmembrane proteins in the protein data bank. Nucleic Acids Res. 33(Database issue), D275-278.
website: http://pdbtm.enzim.hu
For OPM:
Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI (2006) OPM: Orientations of Proteins in Membranes database. Bioinformatics 22, 623-625.
website: http://opm.phar.umich.edu
For PPM Server:
Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL (2011) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res., ASAP online since September 2, 2011.
website: http://opm.phar.umich.edu/server.php
The topologies of transmembrane proteins follow MPtopo:
Jayasinghe S, Hristova K, and White SH (2001) MPtopo: A database of membrane protein topology Protein Science 10: 455-458.
For CmME:
Sommer B, Dingersen D, Gamroth C, Schneider SE, Rubert S, Krüger J, Dietz, K-D: CELLmicrocosmos 2.2 MembraneEditor (2011): A Modular Interactive Shape-Based Software Approach To Solve Heterogeneous Membrane Packing Problems. J. Chem. Inf. Model. 51(5), 1165-1182.
website: http://Cm2.CELLmicrocosmos.org
Aligning a Sample Lipid
A newly added sample lipid has most likely been already aligned the way it should be by the application. If not, or if you have different plans, you are free to change its alignment.
By aligning a sample lipid, you define its default rotation that the membrane algorithms will address to when building your membrane. Basically, you should be aiming at rotating them in a way that they are "standing upright" (extracellular side) or "upside down" (intracellular side).
In order to start, select a sample lipid from the list in the Component View and press the rotate button  .
.

Rotate the lipid by using the mouse. A vertical line will support your orientation.
Again you can find some buttons in the lower toolbar:
Auto Rotation
Automatically rotates the lipid to an upright alignment
Reset
Resets your new alignment to the previous values
Finishing or Canceling
Finish by pressing the accept button . If any similar lipid template has been added to the opposite membrane side, you will be asked if you also wish to apply your alignment, turned to 180 degrees.
To cancel, press the cancel button .
- Details
- Written by bjoern
- Category: Cm2help 5. Building a Membrane
- Published: 14 September 2013
- Hits: 10487
 Cm2 MembraneEditor
Cm2 MembraneEditor  project ...
project ...
 help
help