
7.2. Predefine Conformation
Activate the right tab in order to switch to the conformation mode.
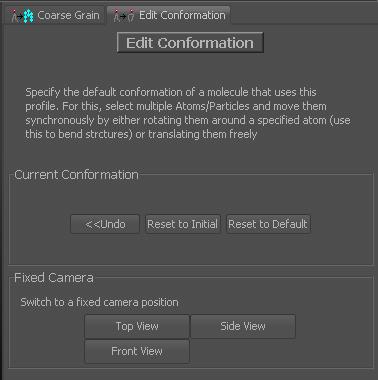
On the right panel, you will find some major options. You can either undo several of the changes, which you may need to use quite frequently. You can also restore the initial conformation, as it was when entering the conformation mode. You can also reset the conformation to PDB file defaults. Another couple of buttons can be used to reset the camera to some fixed positions.
Editing the conformation
Before you can change anything, you'll have to pick one or more atoms that you want to move. You can select/deselect single atoms by left-clicking on them. A double left-click into the background deselects all. Another option is to use rubberband selection. Hold down the ALT-Key in order to draw a rubberband and release it afterwards. Use the left mouse to rotate the camera in the meantime, to reach all of your requested atoms.
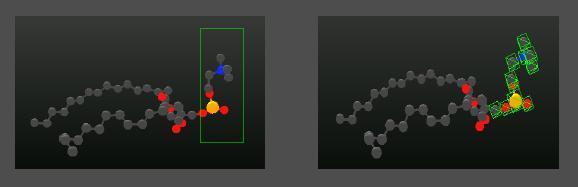
Example of a selection performed by rubberband
There are two different ways to move atoms:
- Rotating them around another chosen atom that serves as a rotation point. A rotation point can be selected/deselected using the right mouse button. Rotate the atoms around this center point by moving the mouse while holding down the SHIFT-Key. You may release the SHIFT-Key in order to change the point of view and resume the process at any time.
The advantage of rotation is that bond length remains unchanged, assuming that you have only picked a group of neighboring atoms. - Translating them. This can be achieved by holding down the CTRL-Key while moving the mouse. Translation will affect bond lengths which will be displayed for a better overview.
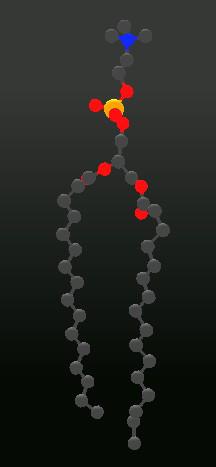
The following image shows an example of how the right carbon-tail of a lipid could be elongated by using a stepwise combination of
picking atoms -> picking a rotation center -> rotating :

After a little more work, this lipid could look like this:
- Details
- Written by bjoern
- Category: Cm2help 7. Atomic Representation
- Published: 14 September 2013
- Hits: 11972
 Cm2 MembraneEditor
Cm2 MembraneEditor  project ...
project ...
 help
help